近年来,蛋白质相关数据海量增加,极大地促进了蛋白质组学的发展,高通量蛋白质组学数据挖掘研究已成为国内外生物信息学研究的热点和学科前沿。蛋白质的功能、代谢以及信号传导等生物过程都与其亚细胞定位密切相关,新合成的蛋白质必须处于合适的亚细胞位置才能正常行使其功能。而异常的蛋白质亚细胞位置能够影响蛋白质的功能,并与人类疾病息息相关,如阿尔兹海默症、肝脏肿瘤、乳腺癌、小唾液腺肿瘤、肾结石和巴特综合征等。研究发现越来越多的蛋白质属于两个或多个亚细胞位置,这些多标记蛋白质通常具有复杂的合成、排列和代谢机制,具有特殊的生物学功能。随着高通量蛋白质测序技术的发展,通过实验方法识别多标记蛋白质已远远不能满足研究的需要,因此发展快速高效的人工智能方法对多标记蛋白质亚细胞位置的精准预测仍是生物信息学的一项挑战任务。
近日,青岛科技大学数理学院人工智能与生物医学大数据研究团队于彬副教授,在生物信息学顶级期刊Briefings in Bioinformatics (IF=8.990)上发表题为“Accurate prediction of multi-label protein subcellular localization through multi-view feature learning with RBRL classifier”的研究论文。报道了构建预测多标记蛋白质亚细胞位置的人工智能模型—Mps-mvRBRL。该模型表现出较强的鲁棒性和泛化能力。于彬副教授为论文的通讯作者,研究生张琪、副教授张艳丹并列第一作者,青岛科技大学为第一完成单位。
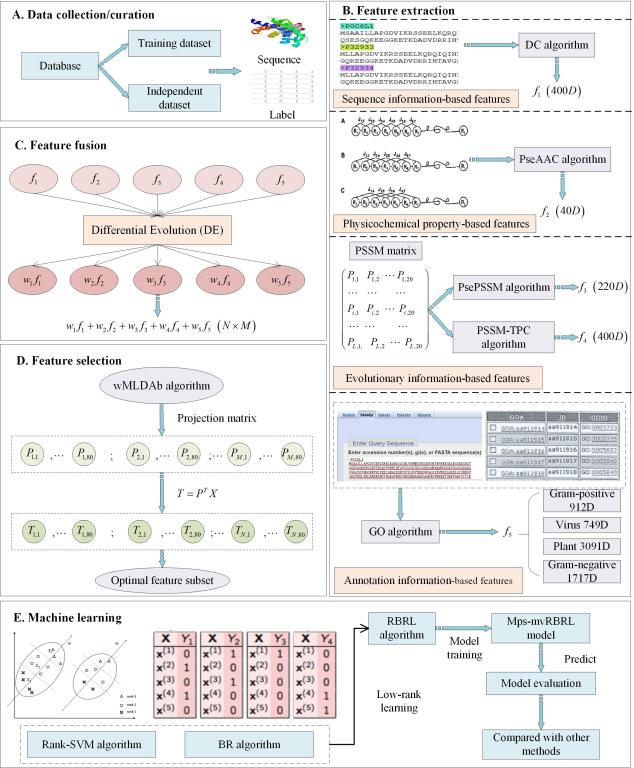
本研究摒弃传统首尾相接的融合方式,根据单特征编码算法的性质,首次引入差分进化算法学习五种单特征的权重向量,通过加权形式融合多视图信息。将融合后的高维特征使用基于二进制权重形式的加权多标签线性判别分析法(wMLDAb)去除冗余信息。此外,本研究首次使用由Rank-SVM和BR算法结合低秩学习(RBRL)算法预测多标记蛋白质亚细胞的位置。通过最严格的留一法检验,Mps-mvRBRL预测模型在革兰氏阴性菌、革兰氏阳性菌、病毒及植物数据集均优于其它已报道的先进预测模型。且Mps-mvRBRL模型的时间复杂度低,具有优异的预测能力和计算稳定性。Mps-mvRBRL模型能够准确预测多标记蛋白质亚细胞位置,并能够应用到更多的多标记蛋白质属性预测中。通过对多标记蛋白质亚细胞位置的预测研究,为确定蛋白质功能提供重要线索,有助于了解蛋白质之间相互作用和调控机制,对某些疾病的发病机理和新药研发具有重要意义。
文章链接:https://doi.org/10.1093/bib/bbab012
Briefings in Bioinformatics是牛津大学出版社(Oxford Academic)出版的JCR一区顶级期刊,2020年的影响因子为8.990,在SCI收录的59个“Mathematical & Computational Biology”类期刊中排名第1,在SCI收录的79个“Biochemical Research Methods”类期刊中排名第3。






